Vorlage Methodenvalidierungsplan Identifizierungsmethode

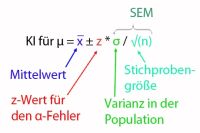
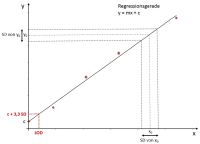
Wenn im Pharmabereich in einem Labor eine Methode, die zukünftig zur Qualitätskontrolle von Arzneimitteln eingesetzt werden soll, (neu) validiert werden soll, ist dafür die Erstellung eines Validierungsplans für die Methodenvalidierung erforderlich. Dabei sind die regulatorischen Anforderungen, wie der ICH Q2(R1), zu berücksichtigen. Ein klar strukturierter und verständlicher Aufbau eines Validierungsplans erleichtert die spätere Durchführung der Validierung der analytischen Methode.