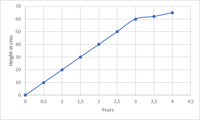
Was ist die ICH Q2(R2) Richtlinie?

Die Richtlinie mit dem Titel „Validation of Analytical Procedures" (ICH Q2(R2)) wurde von der Internationalen Konferenz zur Harmonisierung der technischen Anforderungen für die Registrierung von Humanarzneimitteln (ICH) veröffentlicht. Sie enthält Validierungsparameter für eine Vielzahl analytischer Methoden und wurde zum Zwecke der Harmonisierung (also der Angleichung der Anforderungen) beim Zulassungsverfahren von Arzneimitteln für verschiedene Märkte herausgegeben. Die drei an dieser Harmonisierung beteiligten Parteien sind die USA, die EU und Japan. Es gibt eine ganze Reihe weiterer ICH-Richtlinien, die in 4 Hauptgruppen eingeteilt sind: