Das Leben eines Arzneimittels
In diesem Artikel werden wir über den Lebenszyklus eines Medikaments von seiner Entdeckung bis zu seiner Markteinführung mit Fokus auf seiner Entwicklung und Validierung schreiben.
Arzneimittelentdeckung und -entwicklung
Jedes Arzneimittel, das auf dem Markt verkauft wird, beginnt seine Reise mit seiner Entdeckung. Forscher verbringen Jahre damit, einen Wirkstoff für eine bestimmte Zielsetzung zu identifizieren.
Im Allgemeinen wird ein Medikament aufgrund neuer wissenschaftlicher Erkenntnisse identifiziert, die Wissenschaftlern das Wissen vermitteln, das Fortschreiten einer Krankheit zu verlangsamen oder zu stoppen, oder weil bestehende Medikamente unerwünschte Nebenwirkungen zeigen oder dank neuer fortschrittlicher Technologien, die dabei helfen, einen besseren Kandidaten für das gegebene Ziel zu finden. Die Identifizierung geeigneter Kandidaten erfordert das Screening vieler potenzieller Kandidaten und deren anschließende Eingrenzung auf das am besten geeignete Molekül (Abbildung 1).
Abbildung 1: Ein Pyramidendiagram veranschaulicht die Selektion des finalen Kandidaten aus einem Pool potentieller Wirkstoffkandidaten.
Sobald ein geeigneter Kandidat identifiziert worden ist, folgt dessen Entwicklungsphase. Während dieser Phase wird das zukünftige Medikament strengen Tests unterzogen, da dies die Phase ist, die seinen weiteren Verlauf bestimmt. So ist es u.a. wichtig, sein Absorptionsmuster und seine metabolischen Eigenschaften zu verstehen. In dieser Phase werden unter anderem seine Vorteile, sein Wirkmechanismus und seine Darreichungsform identifiziert.
Testmethoden, Methodenvalidierung und Verifizierung
Während der Entwicklung (sowie für die spätere Freigabe- und Stabilitätsanalytik) werden von den Pharmaunternehmen unterschiedliche Testmethoden (oder auch Prüfmethoden) eingesetzt. Viele Unternehmen lagern diesen Schritt an spezialisierte Auftragslabore (Contract Research Organizations, CRO) oder Lohnhersteller (Contract Manufacturing Organizations, CMO) und ihre zugehörigen Labore aus. Diese Organisationen arbeiten unter den vorgegebenen Good Manufacturing Practices (GMP)-Bedingungen und sind darauf spezialisiert, das Pharmaunternehmen bei der Arzneimittelentwicklung zu unterstützen und die potenziellen Prototypen nach der Etablierung geeigneter Analysemethoden zu untersuchen.
Gemäß der ICH Q2(R2)-Richtlinie werden analytische Methoden hauptsächlich (aber nicht ausschließlich) in Methoden für
- die Identifizierung des Wirkstoffs,
- die Bestimmung von Verunreinigungen (Reinheitstests) und
- die Bestimmung von Gehalt und Aktivität des Wirkstoffs
eingeteilt. Das Labor, das die geeigneten Methoden zur Analyse entwickelt, muss sich anschließend darauf vorbereiten, die Methoden nach deren Etablierung zu validieren. Die Validierung einer analytischen Methode ist ein komplexes Unterfangen. Es erfordert die Erfüllung definierter Akzeptanzkriterien, die für jeden der Leistungscharakteristika und Validierungstests festzulegen sind. Eine solche Methodenvalidierung ist wichtig, weil sie beweist, dass die analytische Methode auch tatsächlich zur Analyse des Analyten geeignet ist und belegt, dass die gemachten Angaben der Wahrheit entsprechen.
Neben der Palette der hauseigenen Analysemethoden sind weitere Prüfmethoden zu berücksichtigen, die die Eignung des Medikaments für seine Verwendung und seine durchgängige Qualität belegen. Dies sind die in den Arzneibüchern (Pharmakopöen) aufgeführten Arzneibuchmethoden. Ein Beispiel ist der bakterielle Endotoxintest des fertigen Produkts (z.B. als LAL-Test). Bekanntermaßen rufen bakterielle Endotoxine u.a. Fieber hervor. Der Endotoxintest des Arzneimittels ist wichtig, um sicherzustellen, dass das Produkt frei von jeglicher Endotoxinkontamination ist. Der Endotoxintest wird oft als präklinischer Test angesehen, ist aber auch regulärer Bestandteil der Freigabemethoden, die für jede Charge anzuwenden sind, sobald das Medikament von den Behörden zugelassen ist und somit auf den Markt gebracht werden kann. Obwohl er kein Parameter des eigentlichen Produktverhaltens im menschlichen Körper ist, entspricht seine Untersuchung der absoluten Grundfrage der Patientensicherheit und muss daher unbedingt durchgeführt werden. Analog der Validierung der selbst entwickelten Methoden müssen die Arzneibuchmethoden verifiziert werden, um ihre Anwendbarkeit unter den aktuellen Bedingungen des Labors zu zeigen, das diese Methode anwenden soll.
Spezifikationen und Qualitätskontrolle
Einen kurzen Abriss zu Spezifikationen gibt dieser Artikel.
Dossiereinreichung, Produktion und Markteinführung
Kehren wir zum Lebenszyklus des Arzneimittels zurück und setzen wir den Entwicklungsprozess fort. Sobald alle präklinischen und klinischen Studien erfolgreich abgeschlossen, alle Spezifikationen festgelegt, alle Methoden etabliert und validiert worden und alle Validierungschargen hergestellt sind, kann das Pharmaunternehmen alle erhaltenen Ergebnisse als Dossier an die jeweilige(n) Zulassungsbehörde(n) übermitteln und die Marktzulassung beantragen. Im Zuge der Zulassung sind Unterlagen für die folgenden Punkte erforderlich:
- Qualität: darunter fallen u.a. die Nachweise zur Reinheit und Stabilität des Arzneimittels
- Wirksamkeit: hierzu zählen die Unterlagen der präklinischen und klinischen Studien, die belegen, dass das Arzneimittel seinen vorgesehenen Zweck erfüllt
- Unbedenklichkeit: hierzu zählen die toxikologischen Untersuchungen, welche möglichst geringe bzw. vertretbare Nebenwirkungen aufzeigen.
Nach erfolgter Zulassung stellt das Unternehmen das Produkt gemäß Marktnachfrage her. Jeder Produktionszyklus führt zu einer Charge. Nach der Produktion wird jede dieser Chargen einer Qualitätskontrolle unterzogen, bei dem die in der Spezifikation definierten Freigabemethoden angewandt werden. Anschließend werden das Analysenzertifikat und der Herstellungsanweisungsbericht (HAB) von der QA-Abteilung, insbesondere von der sachkundigen Person (qualified person, QP), überprüft. Im Falle keiner Beanstandungen und vollständiger Einhaltung aller GMP-Anforderungen wird die Charge für den Markt freigegeben. Neben der Freigabe durch den Hersteller erfolgt für einige besondere Arzneimittel (wie z.B. Impfstoffe, Allergenen, aus Blutplasma hergestellten Produkten u.a.), die in Deutschland in Verkehr gebracht werden sollen, außerdem eine zusätzliche staatliche Chargenprüfung und Freigabe durch das Paul-Ehrlich-Institut gemäß § 32 des Arzneimittelgesetzes (AMG).
Die Europäische Union (EU) hat zum Beispiel sehr strenge Vorschriften für Arzneimittel, die aus einem Drittland in die EU importiert werden. In solchen Fällen müssen die freigegebenen Chargen erneut getestet werden und erfordern verschiedene Dokumente wie eine Importlizenz, ein GMP-Zertifikat und eine Zollbescheinigung. Die Länder mit einem gegenseitigen Anerkennungsabkommen (Mutual Recognition Agreement, MRA) benötigen keinen erneuten Test, allerdings ist ein Chargenzertifikat erforderlich. Ein allgemeiner Ablauf ist in Abbildung 2 dargestellt.
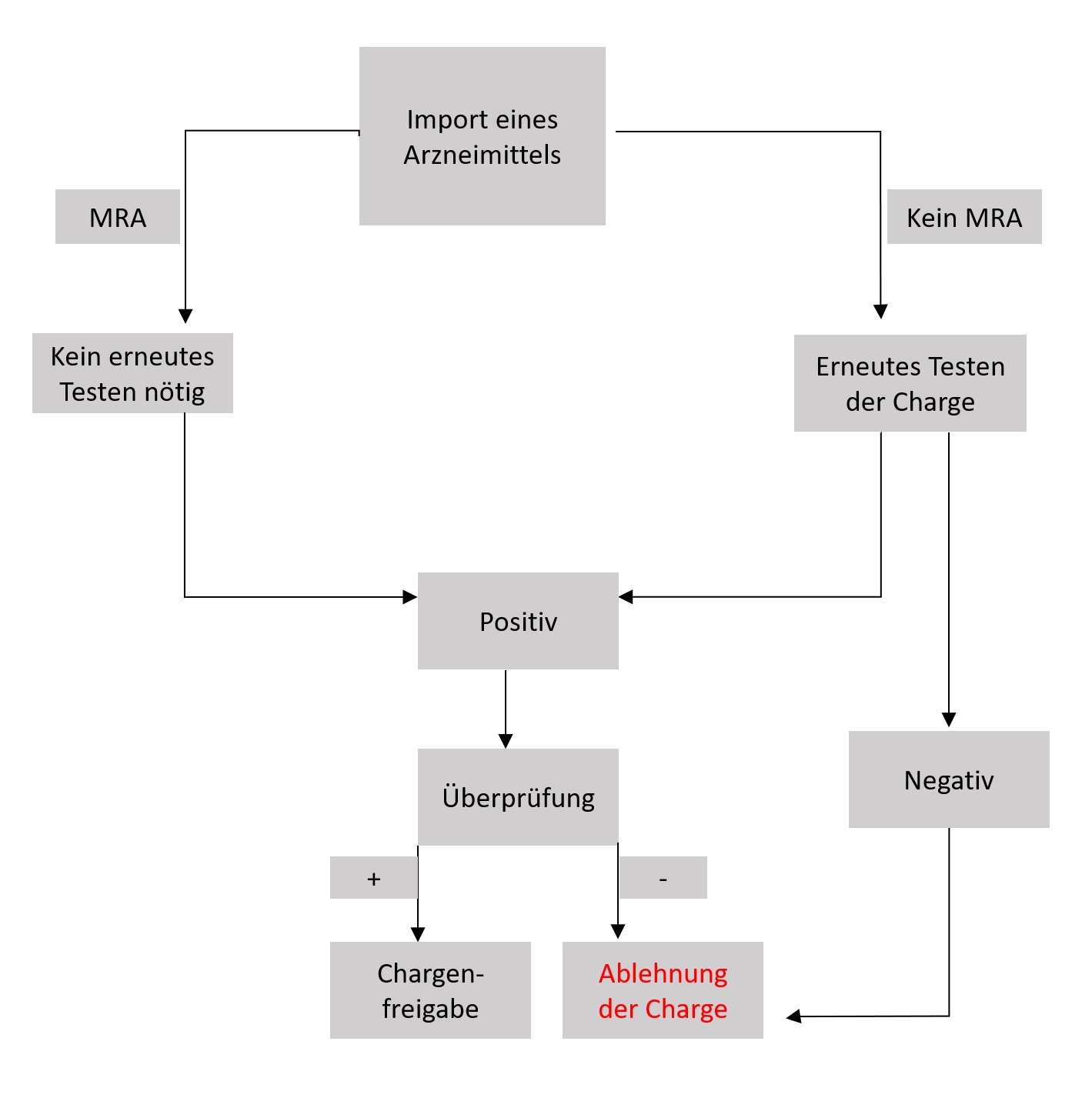
Abbildung 2: Ein Flowchart veranschaulicht den Freigabe-Prozess einer importierten Arzneimittelcharge.
Natürlich resultiert jede fehlgeschlagene oder abgelehnte Charge in verlorenem Geld und Zeit. Daher sollte die Einhaltung der GMP-Anforderungen und die Herstellung einer guten Qualität für jedes pharmazeutische Unternehmen obligatorisch sein.