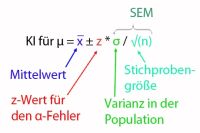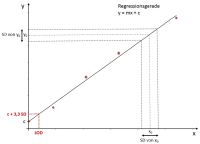
Für die Freigabe von Arzneimitteln ist nicht nur entscheidend, dass der Wirkstoff in angegebener Konzentration und Wirksamkeit vorhanden ist, sondern auch, dass das Arzneimittel möglichst wenig Verunreinigungen aufweist. Solche Verunreinigungen können z.B. herstellungsbedingt sein. Ein Beispiel ist die biotechnologische Herstellung des Arzneimittels (z.B. eines therapeutischen Proteins) mit Hilfe von Bakterien. Bei sterilen, z.B. parenteral verabreichten Arzneimitteln sollten nicht nur keine Keime mehr im Produkt vorhanden sein, sondern auch keine bakteriellen „Rückstände“. Darunter sind z.B. fieberauslösende Bestandteile der Zellmembran bestimmter Bakterien zu verstehen, sogenannte Endotoxine. Diese können mit verschiedenen Methoden nachgewiesen werden, eine davon ist der Limulus-Amöbozyten-Lysat (LAL) Test.