What is a validation plan and what does it tell us?
Method validation is a confirmatory process for the analytical procedures employed, to assess their suitability for intended use. Validation results are used to evaluate the quality, reliability and consistency of analytical methods; it is hence considered an integral part of any good analytical practice and an obligatory prerequisite for every method to be used in a pharmaceutical QC lab or associated CROs (contract research organizations) for the analysis of drugs under GMP conditions.
As a rule of thumb, analytical methods are required to be validated or re-validated
- before their introduction into routine use for analytical purposes;
- for any changes occurring for which the method was initially validated (e.g. change in buffer content, change in instrument profile, use of a standard from a different supplier, etc.); and
- for any change that is outside the original scope of the method (e.g. extension of the application, if e.g. not only drug substance samples but also previous process intermediates should be analysed).
Every validation consists of a detailed validation plan and a validation report. The validation plan is a document that clearly states the entire plan (in great detail) for the validation whereas the validation report is the document that maps the correctness and deviations of the test from what is described in the plan through performed experiments and obtained results and provides information about the success of the validation. In this blog article, we will speak about the validation plan in detail.
The content of a method validation plan
Writing a validation plan requires tremendous attention to detail to ensure development of a realistic and balance schedule (among others). A good method validation plan should contain the following chapters:
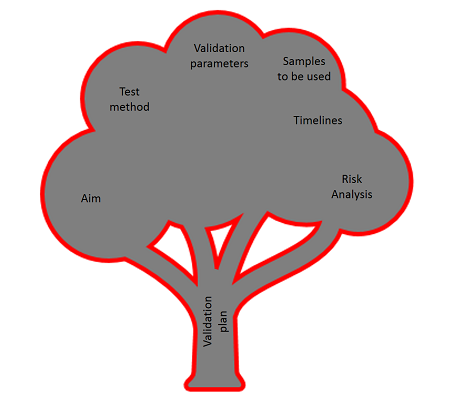
This is not a must, but a recommendation, and as no regulatory requirements exist, the order can also be changed or chapters can be added, omitted, or renamed...
1. Aim
The aim of the plan is similar to an introduction to the document. This usually contains an overview of the method. It starts with listing the intended use of the method, such as e.g. a purity test for DS / DP batch release. This information can be followed by a short description of the method or instruments to be used and some information about the training of the analysts. A validation plan must detail out the parameters that will be determined during the validation and their acceptance criteria (this can be done e.g. by an overview table at the beginning of the validation plan). It should also include the actions required to be taken in case of deviation from original plan or some advice to an appropriate standard operation procedure (SOP) dealing with deviation management.
2. Test Method
In this chapter, a more detailed focus on the analytical method is provided. Here the scope of the method along with details of the used equipment including those for sample preparation, if necessary (instrument model, characteristics), functional details (wavelengths, pH), reagents to be used, assessment details (% peak area, S/N ratio, example chromatograms if appropriate) and information regarding calculations could be defined and an exact description of the analytical performance is given. This might also be very short with just a note to the SOP of the corresponding test instruction (--> document reference) and maybe an explanation of the test / reaction principle. It goes without saying that before starting a method validation, the method to be validated must, of course, be completely described as well as all equipment must be qualified / calibrated 😉
3. Timelines and responsibilities
Here, a realistic timeline for each step of the process along with the respective responsible department or person is listed. Since validation can be a complex process that can include multitude of departments (e.g. in case of co-validations during transfers, like QC, QA etc. of different sites), it is important to consider the schedule of all the involved department or personnel and then map the timelines.
4. Test samples and reference materials
As the header suggests, this section must contain detailed information about the analytes and reference materials. For both, reference standards and analytes, the source (e.g. the lot to be used), the (protein) content, formulation buffer and matrix composition can be clearly stated. Additionally, a clear message about the storage conditions can be provided. Furthermore, for the reference standard its type (primary or secondary standard, international standard, Ph. Eur. reference standard, chemical reference substance, biological reference preparation) can be, if necessary, with a certificate and specification.
5. Validation parameters
This section is of greatest importance as it contains details about all the parameters, such as linearity, precision, trueness, robustness, LOQ / LOD, specificity, and range, that will be determined during the validation according to what is required for that specific method. Not for every type of method every parameter is required, details can be found e.g. in the validation guideline ICH Q2(R1). Along with the listing, this also contains every minute detail about each parameter. For example, how the precision will be demonstrated? Is it only through repeatability, intermediate precision, reproducibility or all of them? For each of these, number of samples, number of analysts, period, instruments used and also the concentration of the analyte / reference standard must be clearly defined. Of crucial importance in this section is the specification of the acceptance criteria for the results of each validation parameter under study. They can be used to evaluate the success of the validation. It is self-evident that acceptance criteria must be defined in advance to have an objective basis for decision-making. If the results obtained are considered without previously defined acceptance criteria, there would be a risk of subjectively judging the results obtained as "somehow fitting", which is not acceptable at all. It should also be stated how the results would be evaluated. In most cases, the obtained data is analyzed statistically (mean, standard deviation, relative standard deviation, etc.) but in some cases such LOD / LOQ, the results can be obtained in multiple ways like S/N ratio, visual assessment etc. Thus, a short explanation and / or e.g. a chapter showing the formulas can be very useful (as e.g. mean and weighed mean should not be mixed up). Because of variability among the scope of the methods, the ICH Q2(R1) has clearly defined the criteria for each kind of method. The tests are classified into three categories: identity, purity, and assay (content and potency). Depending on the method category, different parameters are required to demonstrate the suitability of the method for its intended use.
6. Risk analysis
This chapter chalks out potential risks that may occur during the performance of the analytical test. For example, poor peak resolution due to increase in column temperature. A risk analysis can be a good tool defining the experiments to be done for the evaluation of robustness (if not yet assessed during method development) as it can highlight the critical steps of the method. This chapter is not absolutely necessary, but it is often useful in connection with robustness.
The other sections can include a description about all calculations, if necessary, a glossary, references used, supporting documents, and appendices that may contain all supplementary information. Information regarding the type and scope of the validation report as well as raw data storage should also be given.
In short, a validation plan must be drafted in a way that it contains answer to all the following questions:
- What is the identity of the analyte?
- What are the compositions and handling instructions of the analyte, reference standard, and sample matrix?
- For how long will the validation process run?
- What kind of instruments and inventories will be used?
- Which personnel is involved?
- Where are the samples sourced from and how are they stored?
- What is the scope of the validation?
- What kind of validation parameters will be assessed?
- What are the defined criteria for a successful validation?
- How the results will be assessed?
- In case a problem arises, who or how will it be addressed?
It is also important to note that once the validation plan is signed, any variation / deviation is extremely difficult. Hence, it is advisable to have finished with method optimization (and maybe have performed a method qualification) before planning the validation to determine the possible criteria and to define them in the validation plan some time before starting of the validation activities. Finally, the analysts must be trained in the approved validation plan prior to starting validation activities.
Examples
You can find some example validation plans here for an identity test, a purity determination, and an assay. There is also an example of a superordinate method validation plan that contains the parameters of all three method types and thus enables the selection of the best wording.