What is method validation?
Validating an analytical method proves that by performing the method reliable and reproducible results will be generated, irrespective of time and person who performs. It proves that the method is suitable for its intended purpose.
When and why?
A method validation is necessary when a new method has been established and is supposed to become a routinely used measurement, when aspects of the practical performance have been changed or the method is intended to be used at a different site. This is the case with method transfers. Furthermore, all analytical methods that are planned to be used in a pharmaceutical laboratory for quality control, as well as those used for cleaning validation or environmental monitoring, must be validated prior first application. The requirement to validate the applied analytical test methods is specified in Germany in § 14 of the AMWHV and can also be found in Chapter 6 and 1.9 of the EU GMP guidelines.
According to the Aide mémoire AiM 07123101 of the ZLG, sampling as well as sample preparation (e.g. comminution processes of solids or extraction steps) should be part of the analytical validation. Since sample preparation is part of the analytical method, this is absolutely self-evident. Regarding sampling, it is understandable that sampling must be representative. However, in my opinion, this is not an aspect of method validation, although it must undoubtedly be guaranteed.
Regulatory background: the individual validation parameters
There are regulatory requirements and guidelines to be applied to take critical points into account during method validation. The ICH Q2(R1) is one of them. This guideline „Validation of analytical procedures: text and methodology“, published by the International Conference on Harmonisation, offers help with the classification of different analytical methods and names parameters to be evaluated.
Analytical methods, that are used in the quality control lab, are classified according to their purpose into methods for identification, determination of content (assay) and testing for impurities. Different parameters have to be taken into account during method validation in accordance with this classification. The significant parameters are: trueness, precision (in form of repeatability and intermediate precision), specificity, limit of detection (LOD) and limit of quantification (LOQ), linearity, working range and robustness.
But what do these parameters tell us? The following table provides an overview:
| Validation parameter | Provides information about: | Is determined by (e.g.): | Data / statistical parameter: |
| Trueness | Systematic errors | Spiking experiments | Recovery |
| Repeatability | Random errors within one measurement series in one laboratory (short period of time) | Repeated consecutively performed measurements | Mean, standard deviation, relative standard deviation |
| Intermediate precision | Lab internal random errors (e.g. due to different devices, analysts, etc.) | Measurements at e.g. different days | |
| Specificity | Interference factors, influence of excipients | Sample stressing, placebo measurement | n.a. |
| Limit of detection | Lowest measurable amount | Calculation using the signal-to-noise (S/N) ratio or based on the calibration function | Determined limit concentration |
| Limit of quantification | Lowest quantifiable amount | ||
| Linearity | Relationship between measurement signal and concentration | Analysis of different dilutions | Regression line, correlation coefficient, coefficient of determination |
| Working range | Area between the upper and lower limit in which the method is validated | Successful demonstration of linearity, trueness and precision | n.a. |
| Robustness | Susceptibility to disturbance parameters that can occur during future routine measurements | Small variations in method parameters | Relative difference in respect to the normal condition |
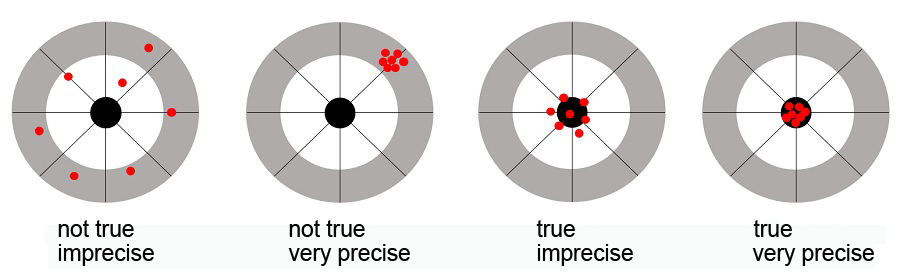
Trueness and precision can be explained with the aid of dartboards. A method can be very precise, all results are close to each other, but unfortunately at the outermost circle of the dartboard. A method can have high trueness, all results hit the bullseye, but unfortunately there’s a great degree of scatter. The results have to hit the bullseye and are where they are supposed to be only when good precision and high trueness have been achieved. The result is reliable.
It’s useful to know the limits of determination and quantification when determining impurities. In this case, the limit of determination for limit tests (transition from “you can’t see anything / you can see something”) and the limit of quantification for quantitative tests are relevant (What is the lowest concentration I can measure quantitatively with a correct result?)
Specificity is essential especially for identity methods. If an identity method is used, it has to be ensured that only the analyte in the sample that is supposed to be determined is detected and that cross-reactions are eliminated. Otherwise, false-positive results are going to be the consequence.
The linearity of the method is of crucial importance for quantitative determinations, where the concentration of the examined analyte is unknown and can fluctuate. A method is linear when the measuring signal is directly proportional to the concentration. The unknown concentration of the analyte can be calculated correctly with the help of a regression line.
The working range of a method can be derived from the linearity, trueness and precision. It’s the linear area between the smallest and biggest concentration in which a true ("accurate") and precise result can be achieved.
A method is robust when it leads to the same result despite smaller deviations from the usual performance procedure. A practical example: There won’t be a difference in the determined molecular weight with a robust SDS-PAGE, even when using precast gels from different suppliers (but with the same concentration). This should be statistically tested with an equivalence test (à comparability) and not, for example, by using an F-test (à differences).
Practical aspects and timing
Prior to the start, it should be checked whether all prerequisites for starting the validation are fulfilled. In addition to the general GMP requirements (such as calibrated and qualified equipment as well as released reagents), this means that the analytical method to be validated has been described at least as a draft in a work or test instruction (e.g. in the form of a SOP) and is also already well established in practice in the laboratory, the analysts are trained in its performance and a signed validation plan is available.
In the validation plan, the procedure for the planned validation is described clearly and unambiguously (i.e. without any scope for interpretation) in detail, with particular reference to the validation parameters to be investigated for the analytical method and the acceptance criteria to be applied. Furthermore, the test material to be used (--> representative sample(s) and reference standard) is specified, among other things. It should be clearly stated what will be done, how, using which sample, by whom and by when. Some information about the content of a method validation plan is given in this blog article and a template for a method validation plan can be found here.
After the analysts have been trained in the validation plan, the practical execution of the experiments takes place and documentation is performed according to the respective company's internal specifications. For example, the validation plan can already be designed in such a way that it includes appendices to be filled in for each individual validation parameter. On the other hand, the general method documentation form can also be completed and the results compiled and evaluated in Excel or Minitab, for example.
The final part is the validation report, a summary, usually written in English, containing the presentation of the individual results, statements on the extent to which the applicable acceptance criteria of the respective parameters were met, and a general statement on whether the validation was successful.
Differentiation from verification and qualification
Method validation must be differentiated from method verification and method qualification. Verification is used for compendial methods, while method validation is applied to in-house developed analysis methods (= non-compendial methods). Although method qualification is also used for in-house methods, it is usually performed earlier and with slightly different intention. Another distinction must be made between the method validation described here and the validation of bioanalytical methods. Details can be found in this blog post.