Was ist GMP?
GMP kommt aus dem Englischen „good manufacturing practice“ und bedeutet auf Deutsch: gute Herstellungspraxis. Damit ist außer einem einwandfreien Herstellungsprozess die Qualitätssicherung und -kontrolle des entstehenden Produkts gemeint. Wichtig ist dies bei Produkten, die am Ende beim / im Verbraucher landen, also in erster Linie Arzneimittel und deren Wirkstoffe, aber in übertragener Form auch Kosmetika, Lebensmittel und Futtermittel.
Wenn bei der Herstellung dieser Produkte etwas schiefgeht, kann sich das also direkt oder indirekt (minderwertiges Futter in der tierischen Erzeugung) auf die Gesundheit vom Verbraucher auswirken. Um dies zu verhindern, wurden Richtlinien zur Einhaltung einer guten Herstellungspraxis entworfen.
GMP und Arzneimittel
Gerade im Bereich der Arzneimittelherstellung ist gute Herstellungspraxis sehr wichtig, da die Gesundheit der Patienten von einer konstant guten Qualität des Arzneimittels abhängt. In diesem Zusammenhang sind zwei Begriffe von entscheidender Bedeutung: die Patientensicherheit und die Produktqualität. Es versteht sich von selbst, dass der Patient durch die Einnahme des Arzneimittels möglichst in keinster Weise geschädigt werden sollte (à Patientensicherheit), gleichzeitig soll durch eine hohe Produktqualität auch der eigentliche Zweck der Heilung / Linderung sichergestellt sein.
Daher wurden für den Vertrieb und die Herstellung von Arzneimitteln besondere Richtlinien und Gesetze erstellt. Hier spielen neben dem Arzneimittelgesetz (AMG) und der Arzneimittel- und Wirkstoffherstellungsverordnung (AMWHV) sowie dem EU-GMP-Leitfaden insbesondere auch die Richtlinien des „International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use“ (ICH), wie beispielsweise die ICH Q2(R2), eine Rolle. Durch diese Richtlinien wird sichergestellt, dass Arzneimittel nicht nur konstant qualitativ gut sind, sondern auch bezüglich ihrer Wirksamkeit und Unbedenklichkeit im Rahmen der Zulassung beurteilt werden. Diese Basisanforderungen Qualität (quality), Wirksamkeit (efficacy) und Unbedenklichkeit (safety) entspringen dem Grundgedanken des AMG: "Es ist der Zweck dieses Gesetzes, im Interesse einer ordnungsgemäßen Arzneimittelversorgung von Mensch und Tier für die Sicherheit im Verkehr mit Arzneimitteln, insbesondere für die Qualität, Wirksamkeit und Unbedenklichkeit der Arzneimittel nach Maßgabe der folgenden Vorschriften zu sorgen." (AMG §1).
Doch kommen wir zurück zur Qualität eines Arzneimittels: Diese hängt schließlich direkt von der guten Herstellungspraxis ab. Um eine gute Herstellungspraxis einhalten zu können, muss auch das Drumherum stimmen. Hierzu gehören neben validierten Methoden zur Herstellung und Qualitätskontrolle auch die geeigneten Räumlichkeiten, qualifizierte Geräte und geschultes Personal. Daher spricht man in diesem Zusammenhang auch von einem Qualitätsmanagementsystem, das all diese Punkte beinhaltet, um die gute Herstellungspraxis zu gewährleisten.
GMP-gerechtes Qualitätsmanagement
Ein Betrieb, der sich an die gute Herstellungspraxis halten möchte, muss sich zunächst um einige Grundlagen kümmern: Die Einführung eines Systems zum Dokumentenmanagement erleichtert die Verwaltung des bald entstehenden Papierkrams – denn zur guten Herstellungspraxis gehört eine ordentliche, lückenlose Dokumentation. Dokumentiert wird so ziemlich alles, was mit dem Herstellungsprozess des Arzneimittels zu tun hat. Dazu gehört die Dokumentation aller Herstellungsprozesse und der darin inbegriffenen Inprozesskontrollen (IPK) und die Kontrolle des fertigen Arzneimittels in der Qualitätskontrolle (quality control, QC), seiner Verpackung und allem Drumherum. Nicht zu vergessen sind auch die Wareneingangskontrollen der Rohstoffe und Primärpackmittel. Sollte es dennoch mal zu Abweichungen im Prozessablauf kommen, müssen auch diese Abweichungen in jedem Fall dokumentiert, anschließend bewertet und Vorbeugemaßnahmen festgelegt werden.
Natürlich muss auch die Hardware stimmen: Alle Geräte und die Räumlichkeiten müssen auf ihre Eignung und Zuverlässigkeit untersucht und dieses natürlich auch dokumentiert werden. Wie wir an diesem kurzen Abriss der Voraussetzungen für die gute Herstellungspraxis sehen, kommt da Einiges zusammen. Daher ist es sehr wichtig, alle beteiligten Mitarbeiter ausreichend zu schulen, damit es nicht zu lästigen Flüchtigkeitsfehlern kommt (die nur noch mehr Papierkram zur Folge hätten). Einfache und klar formulierte Arbeitsanweisungen (SOPs) helfen, Fehler zu vermeiden.
Sind alle Räumlichkeiten qualifiziert, laufen die Geräte und die Dokumentation reibungslos? Dann kann es losgehen – mit der Prozess- und in unserem Falle viel interessanteren Methodenvalidierung!
Die Methodenvalidierung – wozu denn das?
Ganz einfach: Durch den Validierungsvorgang werden die Parameter gesetzt und alle Schritte genau dokumentiert und festgehalten. Die jeweilige Methode wird sozusagen auf Herz und Nieren überprüft. Erst wenn sie diesen „Test“ bestanden hat, kann man guten Gewissens sagen, dass es sich um eine (auch von anderen Mitarbeitern und ggf. anderen Laboratorien) reproduzierbare und zuverlässige Methode handelt, bei der man sich auf das Ergebnis verlassen kann. Nicht nur bei der Herstellung, sondern auch bei der Qualitätskontrolle (während und nach der Fertigstellung eines Arzneimittels) ist ein validierter Prozess bzw. eine validierte Analytik-Methode unerlässlich. Der Einsatz validierter Methoden bei der Qualitätskontrolle ist ein wichtiger Bestandteil der guten Herstellungspraxis. Wenn bei den Ausgangsmaterialien bereits die Qualität zu wünschen übriglässt, kann man sich den Herstellungsprozess schenken. Auch die Inprozesskontrollen, also Qualitätskontrollen an kritischen Schritten während des Herstellungsprozesses, ersparen einem viel Ärger und ermöglichen ein Entdecken potenzieller Abweichungen und damit ggf. einen rechtzeitigen Abbruch. Bei Abweichungen im Herstellungsprozess sowie bei Erhalt nicht spezifikationskonformer Ergebnisse während der Freigabeanalytik oder dem Reißen von Grenzwerten bei Inprozesskontrollen muss eine Fehlersuche eingeleitet werden, um die Ursache zu finden und zukünftig weitere Fehler und größere Verluste zu verhindern. Und am Ende entscheidet die Freigabeanalytik, ob das hergestellte Arzneimittel auf den Markt gebracht werden darf. Dabei erfolgt die Herstellung von Arzneimitteln in sogenannten Chargen, worunter man die Gesamtheit aller in einem Arbeitsgang hergestellten Endbehältnisse (z.B. Fläschchen, sog. Vials, Ampullen oder Spritzen) versteht. Die Freigabenanalytik umfasst dabei einen während der Zulassung des Arzneimittels definierten Umfang verschiedener analytischer Tests, um die Qualität der hergestellten Charge im Hinblick auf Wirksamkeit, Reinheit und Identität zu überprüfen. Dabei kommen je nach hergestelltem Produkt andere Analysemethoden zum Einsatz.
Wo endet die Qualitätssicherung?
Erst wenn alle Analysen der Qualitätskontrolle einer Charge bestanden sind und der Herstellanweisungsbericht (HAB) lückenlos dokumentiert und einwandfrei nachvollziehbar ist, kann diese Charge des Arzneimittels durch die zuständige Person (auch als sachkundige Person / „qualified person“ bezeichnet) zur Abgabe und zum Verkauf freigegeben werden. Die Kontrollen müssen dabei für eine Charge vom Ausgangsmaterial hin zum fertigen Arzneimittel klar nachvollziehbar sein. Mit der Freigabe der Arzneimittelcharge endet jedoch keinesfalls die Qualitätssicherung. Das fertige Arzneimittel muss innerhalb der Haltbarkeitsdauer ohne Qualitätsverlust nachgewiesenermaßen gelagert und transportiert werden können. Hierfür muss der Transportweg gesichert und die Lagermöglichkeit beim Händler machbar sein.
Einen vereinfachten Überblick gibt die folgende Abbildung:
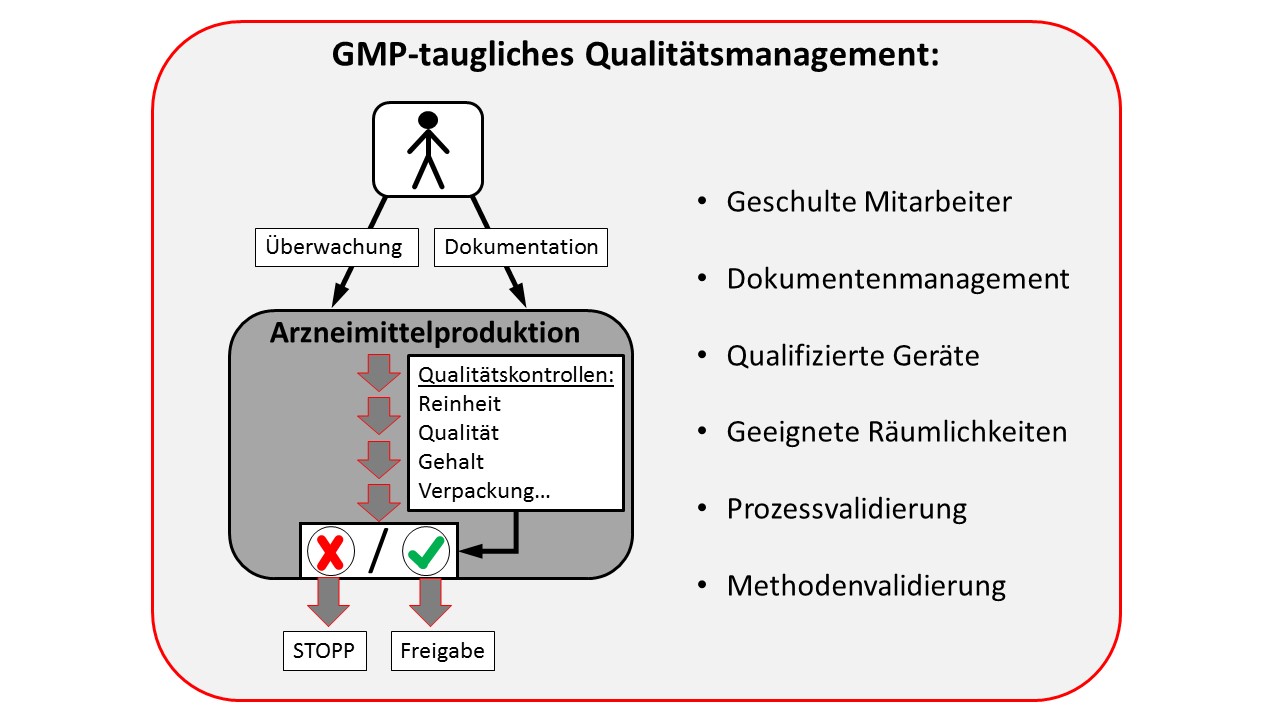
Als Fazit lässt sich sagen, dass die gute Herstellungspraxis durchaus ein schwieriger Weg sein kann – aber nur durch Einhaltung dieser Richtlinien können wir uns als Verbraucher auf die gleichbleibende Qualität der Arzneimittel verlassen.
GMP in der Apotheke
Abschließend wollen wir einen kurzen Ausflug in die Apotheke machen, denn auch bei Arzneimitteln, die in der Apotheke hergestellt werden, wie z.B. einer angerührten Salbe gegen eine Hautkrankheit, muss der GMP-Aspekt berücksichtigt werden.
In diesem Zusammenhang ist zu erwähnen, dass auch für Apotheken ein Qualitätsmanagementsystem erforderlich ist. Dieses soll sicherstellen, dass der Kunde eine gute Beratung erhält und alle in der Apotheke gefertigten Arzneimittel richtig hergestellt, ausreichend geprüft, korrekt gelagert und nicht verwechselt werden können. Dafür sollten die Prozesse analog zur Industrie in Arbeitsanweisungen beschrieben sein. Ein weiterer Aspekt für gute Arzneimittel ist ihre mikrobiologische Qualität. Auf möglichst saubere Herstellungsräume mit einer geringen Keimbelastung zielen die im Hygieneplan festgelegten umzusetzenden Maßnahmen ab.
Genauso wie in der Industrie fallen auch in der Apotheke Prüfungen im Labor an. Dazu zählt beispielsweise die Überprüfung der Identität der einzusetzenden Ausgangsstoffe, was oft mittels Nah-Infrarot (NIR)-Spektroskopie erfolgt. Auch Inprozesskontrollen können anfallen, die bei Rezepturarzneimitteln dann auch bei der Freigabe des hergestellten Arzneimittels durch den Apothekenleiter berücksichtigt werden müssen, da üblicherweise keine Endkontrolle stattfindet.
Neben einer geregelten Lagerung spielt die Dokumentation auch in der Apotheke eine entscheidende Rolle. So ist durch eine schriftliche Herstellungs- und Prüfanweisung festzulegen, wie die Rezeptur oder Defektur anzusetzen bzw. zu prüfen ist und durch auszufüllende Herstellungs- und Prüfungsprotokolle sind die Nachweise der korrekten Umsetzung bzw. die Laborprüfungsergebnisse zu erbringen.