Increasing the sensitivity in case of LOD / LOQ problems in HPLC methods
During validation of purity testing methods of drugs or their active ingredients, sooner or later a point comes when the detection and quantification limits are needed to be determined. The limit of detection or LOD is defined as the lowest amount of an analyte in a sample that can still be detected. This is different from quantification - since the limit of quantitation (LOQ) is the lowest amount of an analyte in a sample that can still be reliably quantified.
This means, the quantification must meet the given requirements in terms of precision and trueness.
LOD and LOQ in HLPC methods
In chromatographic methods such as HPLC (high performance liquid chromatography), these limits are usually determined by means of the signal-to-noise ratio (S/N). The prerequisite for this is the presence of a baseline with baseline noise. The height of this baseline noise results from the halved difference of the lower and upper tangents of the baseline. For a horizontal baseline, this is half the difference between the highest and lowest baseline signals measured (in the figure, upper part). Care should be paid with continuously increasing or decreasing baselines, as often occur in gradient runs. In such cases, it is particularly important to adjust the tangents to the true course of the baseline (see figure below: green lines), so as not to falsely declare a high value as baseline noise (see figure below: red lines). Then, the height of the analyte peak measured from the center of the baseline noise is related to the height of the baseline noise.
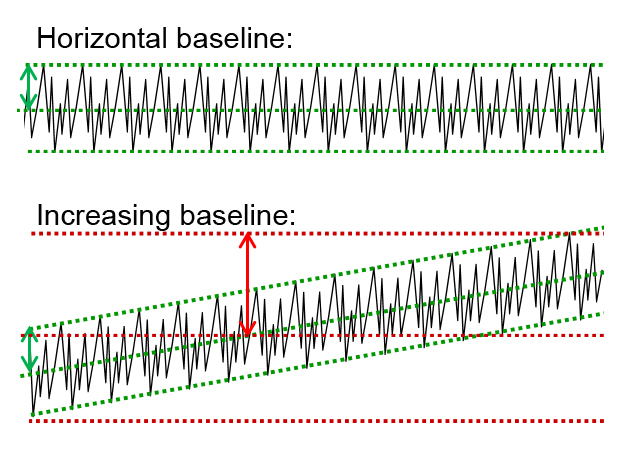
According to the ICH Q2(R2), the harmonized guideline for method validation, at least a signal-to-noise ratio of 2:1 is needed to determine the LOD, better is 3:1. Simply put, if a peak is twice / threetimes as high as the baseline noise, then in most cases it can be reliably identified as a peak. In the case of the LOQ, this is stricter as a ratio of at least 10:1 is suggested as an acceptable value for a reliable quantification.
Solutions for "unchangeable" methods
This poses a particular problem for purity tests: in order to investigate the purity of a product, it is desirable to be able to reliably quantify the lowest possible concentrations of the analyte and, in particular, of the possible impurities. This may mean that even robust routine methods that provide reliable specificity and trueness at the usual working concentrations can fail at low analyte concentrations in terms of LOQ. If it is not allowed to change the method anymore, since it is e.g. has already been submitted to the authority, there are still some ways for corrections. As far as possible, older chromatograms should be used for troubleshooting at this point. If these are showing a lower noise, the focus should be changed to identify the reason behind the increase in noise. First, contamination of the eluents should be investigated. Already a change of the eluent supplier can lead to a clear change of the baseline noise. For aqueous mobile phases, filtration should always be performed and relevant questions such as if the filters are still functioning or if the manufacturer was changed in this case must be answered. Additionally, the degassing of the eluents should always be checked.
Next, the separation column including the guard column must be scrutinized. Even minor impurities can greatly increase baseline noise. Hence, it is best to first use a new guard column and, if necessary, add a new separation column and compare the chromatograms with each other. If the noise is the same, the factor "column" can be cleared of any further suspicions. The next factor to consider could be the HPLC system. It can be checked for any contamination or air in the system. Regular cleaning and maintenance of the system is a prerequisite for good HPLC analysis.
If there is no evidence found from the eluents and the hardware, the cause of the noise can be due to the nature of the method used. If this method and the concentration of the analyte should remain unchanged, there is only one possible approach: The subtraction of the baseline of a blank sample from the analyte sample. If this has not already been done in the validation, this is considered a formal change and must be communicated to the authority, depending on how detailed the dossier has been written. Having the current discussions about data integrity in mind, this approach might not be the first choice and is questionable.
Solution approaches for methods which are still changeable
If changes to the method are still acceptable (for example, due to the current performance of familarization trials during a method transfer), there are other approaches to minimize baseline noise to increase the signal-to-noise ratio. In many cases, the type of solvent used in the eluent is responsible for the noise in the baseline. If the detection of the analyte is done by absorbance measurement, this is particularly significant at low detection wavelengths around 200 nm. First, it should therefore be checked whether the detection can be performed reliably even at higher wavelengths. For example, many substances show a (further) absorbance maximum at 254 nm. If changing the detection wavelength or detection method is not an option, it is worth to look at the eluent selection: methanol displays a higher intrinsic absorption at low wavelengths, which can "swallow" smaller analyte peaks. Although more effort is required, replacement of methanol with acetonitrile may prove worthwhile.
The choice of the separation column can also affect the noise. As a rule of thumb, the smaller the dimensions (especially the diameter) of a column, the higher the peaks appear. This also applies to the entire HPLC system and the capillaries used (see also our blog post on too broad peaks). All peaks are amplified in their height by smaller column / system dimensions, which can also cause a stronger noise. Of course, the peak height still depends on other factors too, such as e.g. the choice of the correct flow rate. A flow rate being too high at a detection frequency being too low may result in losses during detection because the detector simply does not have enough "time" to reasonably detect the analyte. Different column matrices can also affect the sensitivity. It is not possible to provide a general statement on the choice of the right column. In this case it helps to consult different manufacturers and try out samples of different columns.