Die Horwitz-Funktion und ihre Anwendung bei Methodenvalidierungen
Im Zuge einer aktuellen Methodenvalidierung ist mir die „Horwitz-Funktion“ über den Weg gelaufen, welche einen Zusammenhang von Analytkonzentration und beobachteter Messpräzision bei analytischen Methoden aufzeigt. In diesem Blogbeitrag möchte ich ein wenig näher auf ihre Bedeutung eingehen.
Ein wenig Historie und Hintergrund
Diese empirische Beobachtung geht auf 1980 und 1982 veröffentlichte Studien von William Horwitz zurück, einen renommierten ehemaligen Statistiker der FDA [1, 2]. Eine Auswertung von über 150 unabhängigen AOAC-Ringversuchen mit verschiedensten Laboren und Analysemethoden für unterschiedliche Anwendungsbereiche (Pharmazeutika, aber vorwiegend Lebensmittelanalytik) hat gezeigt, dass es eine Art „Grundkurve“ der analytischen Präzision gibt: Wenn die Konzentration C eines Analyten um den Faktor 100 abnimmt, verdoppelt sich die relative Standardabweichung der Reproduzierbarkeit RSDR um etwa zwei Größenordnungen [2]. Einfacher gesagt: Wenn die RSDR bei 100%iger Analytkonzentration bei z.B. bei 2% liegt, beträgt sie bei 1%iger Analytkonzentration 4% und bei 0,01%iger Analytkonzentration 8 % usw. …
Wobei wir eigentlich eher vom Massenanteil anstelle von einer Konzentration sprechen sollten…
Diese Gesetzmäßigkeit wird als „Horwitz-Trompete“ bezeichnet, da sie grafisch wie ein Trichter aussieht:
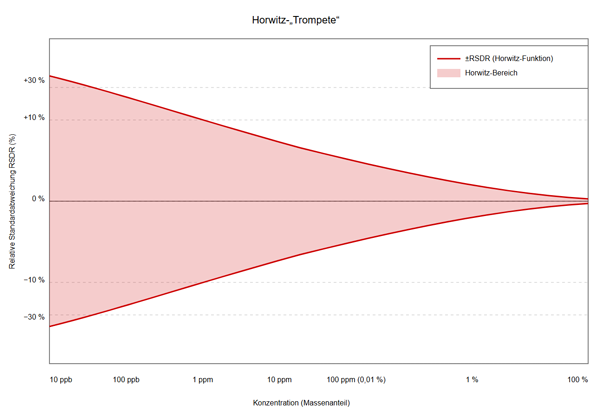
Erstaunlicherweise gilt dieses Muster unabhängig von Substanz, Matrixtyp oder analytischer Methode.
Mathematisch kann dieser Zusammenhang durch eine Potenzfunktion mit einem ungewöhnlichen Exponenten (ca. 0,85) beschrieben werden: RSDR (%) = 2C0.8495 C-1 bzw. 2C-0.1505 [3, 4]. Bei sehr niedrigen Konzentrationen (< 10 ppb) weichen die Ergebnisse jedoch von der klassischen Horwitz-Funktion ab und pendeln sich auf eine relative Standardabweichung von 20 – 25% ein, da sie weitestgehend unterhalb der Nachweisgrenze liegen würden [4]. Daraus lässt sich der „Horwitz Bereich“ von etwa 10-7 – 10-1 Massenanteil ableiten.
Die Anwendung der Horwitz-Funktion
Auch wenn die Horwitz-Funktion für die Lebensmittelanalytik (auf den - laut Aussage des Autors - ihre Anwendung weitgehend beschränkt ist) ein bemerkenswert gutes Vorhersagetool für den Trend der Standardabweichungen der Reproduzierbarkeit im entsprechenden Konzentrationsbereich ist, lässt das nicht den Rückschluss zu, dass sie ohne Weiteres unkritisch für jegliche Validierungen herangezogen werden sollte [5]. Ein aktuelles Paper diesen Jahres zeigt zudem, dass neuere Daten von 20 Ringversuchen in der Lebensmittelanalytik aus den Jahren 2011 bis 2017 nicht dem Horwitz-Model folgen… [6]. In diesem Zusammenhang sei vielleicht auch auf das Paper von Thomson 2012 hingewiesen, welches eine Alternative zur Horwitz-Funktion beschreibt, da diese für einzelne Methoden nur bedingt geeignet sei und Nachweisgrenzen ignoriere [7].
Im Lebensmittel-, Genussmittel-, Veterinär- und Umwelt-Bereich oder in der forensischen Toxikologie findet die Horwitz-Funktion breite Anwendung, z.B. zur Nikotinanalytik in Tabakprodukten, für die unterschiedlichsten Analysemethoden für Wein und Most, für Plastikadditive in Gewässerproben, für Vitamine in Nahrungsergänzungsmitteln, für die Schwermetall- oder Mykotoxinanalytik in Tierfutter oder für Antibiotika in Milch oder Fleisch... Dies ist nur allzu verständlich, da ihre Anwendung dort je nach Anwendungsbereich auch regulatorisch vorgeben ist wie beispielsweise für Pflanzenschutzmittel in der EU-Pestizidleitlinie oder für Lebensmittel über den Codex Alimentarius. Im pharmazeutischen Bereich dagegen ist die Anwendung der Horwitz-Gleichung äußerst rar. Auch wenn es mal eine Erwähnung der Horwitz-Gleichung für die Präzision im USP Medicines Compendium Kapitel <10> gegeben hat, erscheint mir die aktuelle Relevanz für pharmazeutische Methodenvalidierungen recht gering zu sein, denn
- taucht die Horwitz-Funktion in den aktuellen einschlägigen Validierungsrichtlinien und Arzneibuchkapiteln zu Methodenvalidierungen nicht auf und
- wurde auch das USP Medicines Compendium bereits 2015 eingestellt [8, 9].
Obwohl man mit etwas Recherche durchaus ein paar Publikationen zu Methodenvalidierungen und ein White-Paper findet, in denen auf die Horwitz-Funktion Bezug genommen wird [10 – 17], sei das mit der grundsätzlichen Aussagekraft und wirklicher Relevanz im Pharma-Bereich mal dahingestellt…
Die grundsätzliche Beobachtung, dass die Präzision sich verschlechtert, je geringer die Analytkonzentration ist und daher der Gedanke, für Methodenvalidierungen entsprechend breitere Akzeptanzkriterien für geringere Konzentrationen zu wählen (z.B. für eine HPLC-Methode, die gleichzeitig als Gehaltsbestimmung für den Analyten (à 100% Level) und als Impurity-Methode für Verunreinigungen im Spurenbereich (à LOQ / Spezifikationsgrenze)), erscheint sehr verständlich und nachvollziehbar. Aufgrund der begrenzten Datenlage würde ich persönlich die Anwendung des Horwitz-Models für Methodenvalidierungen im pharmazeutischen Bereich dennoch eher nicht empfehlen.
Sollte jemand anderer Meinung sein, hinterlasst mir gerne einen Kommentar – ich freue mich auf die Diskussion 😉
Referenzen
[1] Horwitz W; Kamps LR; Boyer KW, “Quality Assurance in the Analysis of Foods for Trace Constituents”, J. Assoc. Off. Anal. Chem. 1980; 63 (6): 1344-1354
[2] Horwitz W, “Evaluation of Analytical Methods Used for Regulation of Foods and Drugs,” Analytical Chemistry 1982; 54 (1): 67A-76A
[3] Albert R, Horwitz W, “A Heuristic Derivation of the Horwitz Curve”, Anal. Chem. 1997; 69: 789-790
[4] Thompson M, AMC Technical Brief no. 17, (2004) “The amazing Horwitz function”. http://www.rsc.org/pdf/amc/brief17.pdf, zugegriffen am 15.10.2025
[5] Thompson M, “Limitations of the Application of the Horwitz equation: A rebuttal”, Trends in Analytical Chemistry 2007; 26(7): 659-661
[6] Ehling S, Thompson JJ, Schimpf KJ, Pacquette LH, Haselberger PA, “A Contemporary Look at the Precision of Modern Analytical Methods in Food Analysis and the Relevance of the Horwitz Equation”, J AOAC Int. 2025;108(4): 566-571
[7] Thompson M, “The characteristic function, a method-specific alternative to the Horwitz function”, J AOAC Int. 2012; 95(6): 1803-1806
[8] USP Medicines Compendium <10> Assessing Validation Parameters for Reference and Acceptable Procedures - Guideline for Donors / lnstructions for Staff
[9] Schiavetti B, Wynendaele E, De Spiegeleer B, Mbinze GJ, Kalenda N, Marini R, Melotte V, Hasker E, Meessen B, Ravinetto R, Van der Elst J, Mutolo Ngeleka D, “The Quality of Medicines Used in Children and Supplied by Private Pharmaceutical Wholesalers in Kinshasa, Democratic Republic of Congo: A Prospective Survey”. Am J Trop Med Hyg. 2018; 98(3): 894-903
[10] Apostol I, Kelner D, Jiang XG, Huang G, Wypych J, Zhang X, Gastwirt J, Chen K, Fodor S, Hapuarachchi S, Meriage D, Ye F, Poppe L, Szpankowski W, “Uncertainty estimates of purity measurements based on current information: toward a "live validation" of purity methods”, Pharm Res. 2012; 29(12): 3404-3419
[11] Bento D, Borchard G, Gonçalves T, Borges O, “Validation of a new 96-well plate spectrophotometric method for the quantification of compound 48/80 associated with particles”, AAPS PharmSciTech. 2013;14(2): 649-655
[12] Scherer R, Pereira J, Firme J, Lemos M, Lemos M, “Determination of Ciprofloxacin in Pharmaceutical Formulations Using HPLC Method with UV Detection”, Indian J Pharm Sci. 2014;76(6): 541-544
[13] Kumnerdnon P, Rojsitthisak P, Niwattisaiwong N, Sotanaphun U, Chatchawalsaisin J, Sutanthavibul N, “Validation of an RP-HPLC Method for Quantitative Analysis of Phikud Navakot Extract using the Standard Addition Method”, TJPS 2016; 40(1): 26-31
[14] Surasarang S, Karnpracha C, Boonyapiwa B, “Analytical Method Validation for Testing of Limit of High Molecular Weight Proteins in Filgrastim Biopharmaceutical Products”, IJPHS 2019; 1(1): 12-25
[15] Li W, (2019) “Points to Consider in Quality Control Method Validation and Transfer”, BioProcess International; https://www.bioprocessintl.com/qa-qc/points-to-consider-in-quality-control-method-validation-and-transfer, zugegriffen am 15.10.2025
[16] Quynh Trang NT, Van Hop N, Giang Chau ND, Tran TB, “Simultaneous Determination of Amlodipine, Hydrochlorothiazide, and Valsartan in Pharmaceutical Products by a Combination of Full Spectrum Measurement and Kalman Filter Algorithm”, Advances in Materials Science and Engineering 2019; 1-9 (Article ID 5719651)
[17] Marson BM, Concentino V, Junkert AM, Fachi MM, Vilhena RO, Pontarolo R, “Validation of analytical methods in a pharmaceutical quality system: an overview focused on HPLC methods”, Quim. Nova 2020; 43(8): 1190-1203