What is a CAPA process?

CAPA is an abbreviation for "Corrective and Preventive Action". The CAPA process was first introduced in 2006 by the FDA (Food and Drug Administration) in the USA as part of the Quality Systems Guidance issued, which later formed the basis of the ICH Guideline Q10 relevant to the pharmaceutical sector. ICH Guideline Q10 has now also been included in Part 3 of the EU GMP guidelines, highlighting the implementation of the CAPA process in the pharmaceutical environment.
In the field of medical devices, corrective and preventive actions are treated separately in ISO 13485 (chapter 8.5.2: "Corrective Action" and chapter 8.5.3: "Preventive Action"). Therefore, the standard does not explicitly mention the CAPA process as such, but rather divides it into the separate processes CA ("Corrective Action") and PA ("Preventive Action"). The standard requires documentation and evaluation of these improvement and preventive actions. Therefore, the implementation of a CAPA process that combines these two separate processes is a good idea.
A CAPA process is used to deal with non-conformities (deviations or errors) that have occurred (correction) and to prevent future non-conformities (prevention). Here, a distinction must be made between the terms "correction" and "prevention":
- A corrective action always refers to a non-conformity that has already occurred.
- Preventive actions, on the other hand, are exclusively those that are taken before a non-conformity can even occur (for example, adequate labeling for product-related use or the warning signs in the figure above).
The analysis of potential errors, deviations and their consequences can be performed in the course of a risk assessment.
Occurring non-conformities can result, for example, from the evaluation of the management system during an internal audit and from customer feedback or, for companies in the medical device or pharmaceutical industry, from safety-relevant incidents (for example, triggered by defective goods due to inadequate controls in production). A CAPA process initiated by occurring non-conformities therefore starts in any case with those actions and corrections to be carried out immediately in order to keep further effects limited. Depending on the severity, this can be, for example, an immediate production and sales stop in the most critical case.
The actual CAPA process starts after performance of immediate actions and can be compared to the PDCA (Plan-Do-Check-Act) cycle in its execution: It starts with a detailed description of the non-conformity, whereby the problem is precisely evaluated and documented. This step can be equated with the planning phase of the PDCA cycle. Then, after performing a root cause analysis, the necessary corrective actions are determined. These can be, for example, a change in the production process or a replacement of a single component that is important for the completion of the product. Preventive actions are also taken to avoid recurrent occurrence of this non-conformity in the future. Strictly speaking, these are also corrective actions (by definition, pure preventive actions are only those that are perceived before the non-conformity occurs). The defined actions are implemented accordingly after their verification and validation (cf. "Do" phase of the PDCA cycle). At the end, the effectiveness of the actions is checked (effectiveness check) and the success is evaluated accordingly.
In a phase to be compared with the "Act" phase, reasons are given as to whether and why the CAPA process was successful (see figure below).
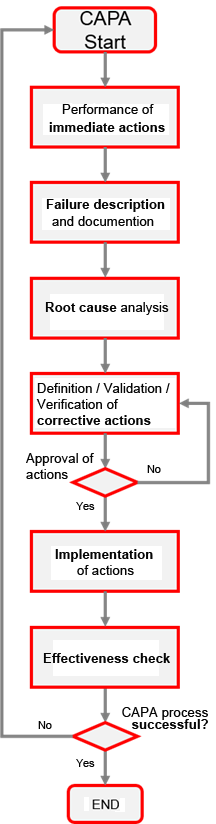
If the CAPA process has not been successful or has only been partially successful, another CAPA process must be initiated. A successfully conducted CAPA process may, under certain circumstances, result in a reassessment or supplementation of the risk analysis.