A drug's life
In this article, we will write about the life cycle of a drug from discovery to its release with a focus on its development and validation.
Drug discovery and development
Every drug, that is sold in the market starts its journey from being discovered. Researchers spend years to identify the drug substance for a particular target.
Generally, a drug is identified because of new scientific updates that provide scientists the knowledge to stop or halt the progression of a disease, existing drugs that show unwanted adverse effects, and advanced technologies that help finding a better candidate for the given target. Identification of suitable candidates requires screening of many potential candidates and then narrowing them to the most fitting compound for the target (Figure 1).
Figure 1: Pyramid diagram to show the selection of the final candidate from a pool of potential drug candidates.
Once the identification is done, the drug is subjected to the development phase. During this phase, it goes through rigorous tests because this is the phase that decides its further progression. I.a. it is important to understand its absorption patterns and metabolizing capabilities. It is in this phase that among others its benefits, mechanism of action and its dosage form are identified.
Test methods, method validation and verification
During development (as well as for subsequent release and stability analysis), different test methods are employed by pharmaceutical companies. Many companies outsource this step to specialised Contract Research Organizations (CRO) or Contract Manufacturing Organizations (CMO) and their associated laboratories. These organizations work under the suggested Good Manufacturing Practices (GMP) environment and are specialized in assisting the pharmaceutical company in drug development and analysing the potential prototypes after establishment of the appropriate analytical procedures.
As per the ICH Q2(R1) guideline, analytical methods are mainly categorized into methods for
- Identification of the drug substance,
- Purity tests, and
- content and potency of the drug substance.
The laboratory that develops the suitable methods for the specific purpose against the distinct drug substance must then prepare to validate the method. Validation of an analytical method is a complex procedure. It requires fulfilling defined acceptance criteria set for each of the validation parameters such as specificity, trueness, linearity, precision, detection and quantification limit, range, and robustness. Such a method validation is important because it proves the that the analytical method is suitable for testing the analyte and shows the authenticity of the claim. Aside the panel of in-house analytical methods there are other test methods as well that prove the suitability of the drug for usage and its consistent quality. These are compendial methods, listed in the pharmacopoeias. One example is the bacterial endotoxin test of the finished product (e.g. the LAL test). Bacterial endotoxins are known to cause fever amongst others. The endotoxin test of the drug is important to ensure that the product is free from any endotoxin contamination. The endotoxin test is often considered as a preclinical test, but also belongs to release testing, once the drug is accepted by the authorities and thus allowed for market release. Although it is not a measure of the product behavior when it is applied to a human body, it answers the absolute basic question of product safety and is thus absolutely required to be demonstrated. Like validation of the developed methods, the compendial methods need to be verified to show their applicability under the current situation in that lab that is intended to use the method.
Specifications and quality control
This article gives a brief overview of specifications.
Dossier submission, production and market release
Let’s return to the drug product life cycle and continue the process of development. Once all preclinical and clinical studies are successfully finished, all specifications set, all methods established and validated, and all validation batches manufactured, the pharmaceutical company may submit all the obtained results as a dossier to the respective regulatory authorities and apply for market authorization (easily said: its acceptance). During application, documents to prove the following aspects are required:
- Quality: this includes i.a. the evidence of purity and stability of the drug
- Potency: this includes the documentation of preclinical and clinical studies demonstrating the drug fulfills its intended purpose
- Safety: this includes toxicological studies demonstrating only small or acceptable adverse effects.
After the application is accepted, the company manufactures the product as per the market demand. Each production cycle is resulting in one lot (or batch). After production, each of these batches is then quality controlled by going through the list of release tests defined in the specification. Then, the CoA and the manufacturing batch record are verified by the QA department, especially by the qualified person (QP), and in case of no negative observations and full compliance to all GMP requirements, the batch is being released into the market. In addition to release testing performed by the manufacturer, for some special pharmaceuticals (such as vaccines, allergens, products made from blood plasma, etc.) that are to be marketed in Germany, an additional governmental batch testing and subsequent batch release is carried out by the Paul Ehrlich Institute in accordance with Section 32 of the German Medicines Act (AMG).
The European Union (EU) for example has very strict regulations for medicinal products that are imported to the EU from a third country. In such cases, the released batches need to be tested again and require different documents such as an import license, a GMP certificate and a customs certificate. The countries with a Mutual Recognition Agreement (MRA) however don’t need any retest but a batch certification is required. A general workflow is shown in figure 2. Of course, every failed or rejected batch results in a lot of lost money and time. Thus, adhering to the GMP requirements and producing good quality should be mandatory for each pharmaceutical company.
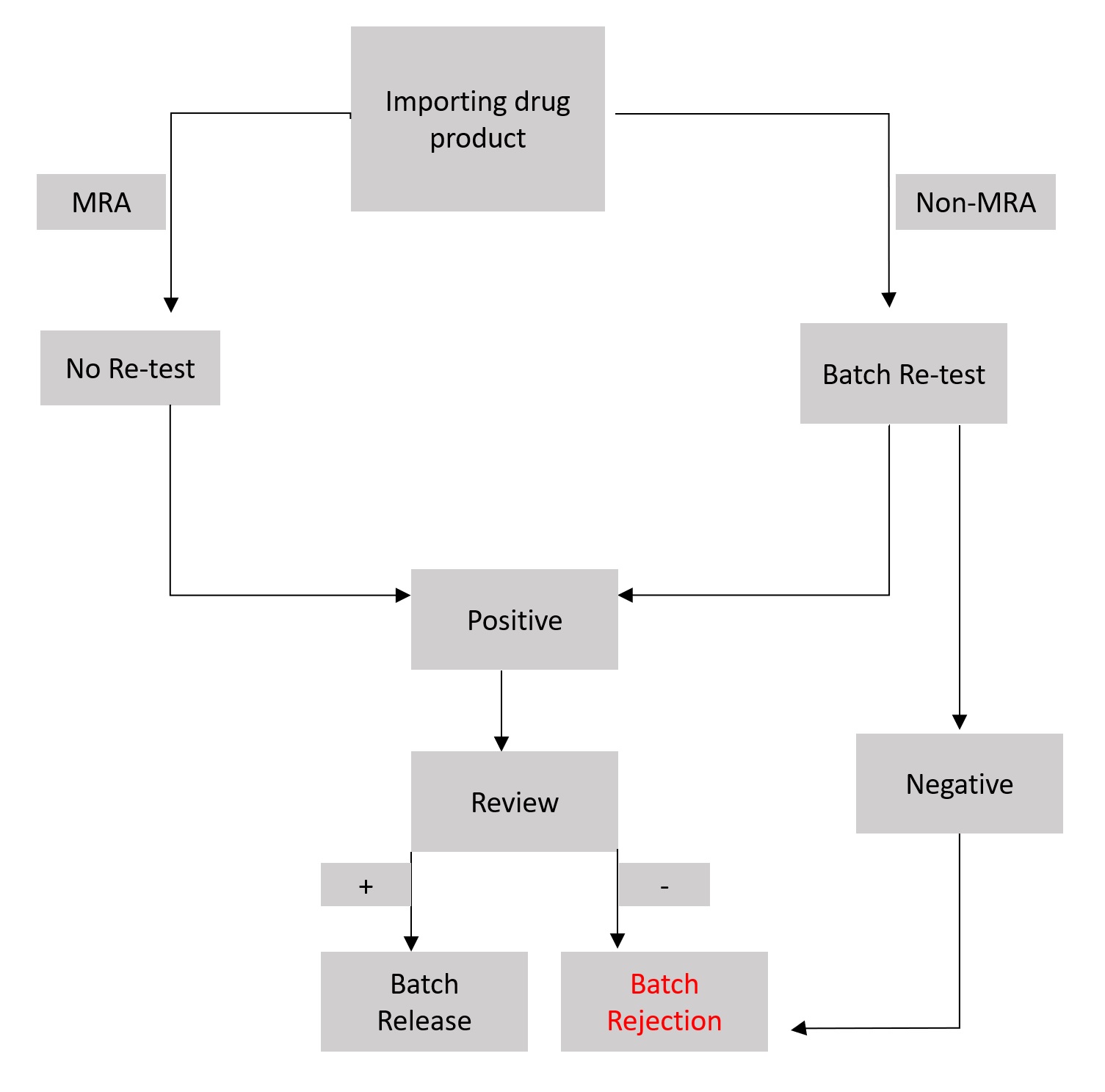
Figure 2: Flowchart explaining the release process of an imported batch of drug product.