HPLC: What to do in case of peaks being too broad?
HPLC methods are often part of routine analysis of drugs or drug substances in pharmaceutical QC laboratories under GMP conditions.
The shape of the peak of a HPLC chromatogram is extremely critical for its evaluation. Subtle changes in the shape during a quantitative measurement can lead to misleading results and in turn can ruin the entire measurements obtained throughout the experiment. Therefore, it is important to keep a watchful eye on the “peak shape”.
This is best done by comparing the recently obtained chromatogram to a pre-existing standard chromatogram at different time points. An abnormal difference in the width or symmetry will lead to discrepancies in the results and requires immediate actions.
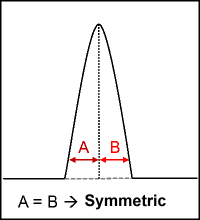
Peak symmetry is easily identified by a simple method. Consider a peak and draw a vertical straight line from the maxima to base of the peak. If the vertical line split the peak in equal halves i.e., distance from the start of the peak to the centre (A) and from centre to the end (B) of the peak is equal (figure 1), it is symmetric, else it is called asymmetric. Possible reasons for asymmetrical peaks will be explained in a separate blog article.
Peaks being too broad are not as easy to identify. A direct comparison of the peaks width with an established standard helps in the identification. But, in case of unavailability of existing standards, especially during the establishment of a new methods, the process could become challenging. First, it must be distinguished whether all peaks have the same width or if individual peaks are significantly wider than others.
What happens when all the peaks appear broader?
Peak widening can happen through various factors and may have “normal” reasons. One can be a flow rate being too low. Each column has an ideal flow rate based on the dimensions of its inner diameter (ID) which is reported by the supplier. Columns with an ID of 5 mm usually use a flow rate of 1 ml/min whereas 3 mm columns have their ideal flow rate set at 0.3 ml/min. Any bigger deviation from the optimal value can lead to a broadening of the peak. Thus, follow the manufacturer’s instruction or try a column with a smaller ID.
Another, often neglected point is the dead volume of the HPLC system. Combining a modern, small-sized column with an older HPLC system, one should consider replacing the capillaries as well. Especially, the diameter of the capillary between the column and the detector has an enormous influence on the peak width - just like an old detector with a flow cell being too large (figure 2). Not only the flow cell but also the parameters of the detector can influence the peak width. Set the data acquisition rate (usually in Hertz) higher and compare the chromatograms. Additionally, it is an absolute must to check the system for leaks. Between the column and the detector even small leaks can have fatal consequences for the quality of the chromatogram.
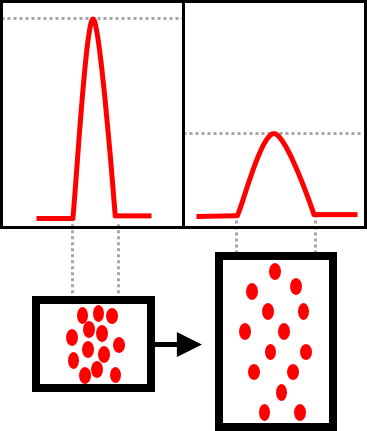
Peaks of one chromatogram may have different widths
Having a chromatogram in which individual peaks appear wider than others may be due to several reasons. While early peaks with a short retention time look narrow, the late peaks with a longer retention time can be very broad. This is normal in most cases. But in certain cases the resolution should be assessed to check whether there are two poorly separated peaks behind one peak being too wide. With the availability of UV / Vis detection, it is worth looking at the absorbance spectrum. If it varies within “one” peak, the presence of two different substances is very certain. Separation of two peaks can be achieved by broadening the gradient or by using another column (e.g. smaller ID, smaller particle size).
If successful, once a broad looking peak could now be resolved into two separate, distinct, and narrow peaks. If this is not the case, you still have the peak being too wide. Before you change all parameters or think about purchasing a new column, let’s talk about possible causes that are often overlooked. Do the peak maxima appear to be cut off? It could be the case of over-shooting the detection limit, thus the detector is out of its linear range. The injection volume may also be too high and “overwhelm” the column (especially in case of a small column ID). In both cases the solution is quite simple: Dilute the sample or reduce the injection volume. The pH of the mobile phase can also have a strong influence on the peak width. Depending on the chemical property of a sample, it can be ionized in different ways, i.e., completely protonated, deprotonated or neutral. The general rule of thumb is that the pH of the mobile phase should never be identical to the pKa of the substance.
If neither a hidden double peak nor an incorrect pH of the mobile phase is the cause of an early, broad peak, then focusing is missing. Adjusting the gradient could be the solution. For example, a classic mistake in reverse phase HPLC is a too high concentration of organic solvent right from the beginning (e.g. to save time). Relatively polar substances are thus carried along from the beginning and are thus detected as broad peaks of low retention time. If the gradient is started with 100% water, these substances will initially remain adhered to the column and can be eluted with increasing solvent concentration in a focused manner.
Broadening of peaks with respect to time
If the width of the peaks changes over time, there could be other reasons apart from the ones already discussed in the previous section. In this case: change the guard column and and if required replace the mobile phase. If the situation does not improve, flushing of the column or a backwash by inverting the column and flushing from back to front, could sort out the issue. A backwash should however be performed as per the manufacturer's instructions, as not all available columns are suitable for backwashing! It is advisable to decouple the detector during longer rinsing operations in order to avoid subsequent contaminations.
Even dirty inline filters, frits, capillaries or injectors (autosampler) can be the problem. If you have a second HPLC system, you can connect the HPLC column in the second system to facilitate the diagnosis. If the chromatogram is normal here, the problem is not related to the column and the first HPLC system has to be thoroughly rinsed and examined for leaks.
If the column also shows too wide peaks in other HPLC systems and if contamination of the HPLC system can be excluded, the HPLC column must be replaced. Depending on the age and number or type of samples this is quite normal. However, if a column replacement happens earlier than anticipated, it may be due to incorrect / missing sample preparation or inappropriately chosen method parameters. A flow rate being too high, too high temperatures, too strong solvents or too high or low pH can accelerate the aging of the column. Always follow the manufacturer's instructions.
Since troubleshooting HPLC is very time consuming, we hope to be able to help with this blog post and have pointed out solution ideas.