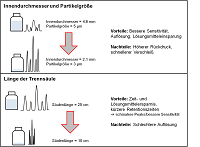
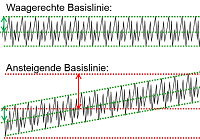
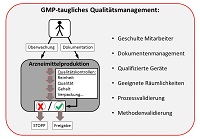
ISO 13485:2016 vs. ISO 9001:2015 - Unterschiede und Gemeinsamkeiten - Teil 2

Im ersten Teil der kleinen Serie zu den Unterschieden und Gemeinsamkeiten zwischen der ISO 13495 und der ISO 9001 bin ich ja auf so grundlegende Dinge eingegangen, wie zum Beispiel die HLS oder die Ausrichtungen der Normen. In diesem Beitrag möchte ich daher genauer auf die einzelnen Normkapitel 4 bis 6 eingehen.