Vorlage zur Berechnung der Linearität bei Methodenvalidierungen

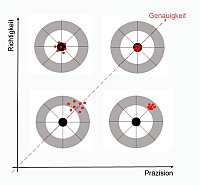
Im Zuge von Methodenvalidierungen in pharmazeutischen QC-Laboren oder beauftragten Lohnlabore fallen unterschiedliche Validierungsparameter an, die es abzuprüfen gilt. So sind z.B. bei quantitativen Bestimmungen von Verunreinigungen und Bestimmungen des Wirkstoffgehalts und der Wirksamkeit Untersuchungen zur Linearität gemäß der Methodenvalidierungsrichtlinie ICH Q2(R1) erforderlich.